
ARTIGOS ORIGINAIS
TUMOR CARCINÓIDE DE RETO
Patrícia Alves Mangueira1, Gabriella Oliveira Fernandes1, Carlúcio Cristino Primo1 , Marco Aurélio Viana França1 , Hilton Pereira Maia 1, José Hermes Gomes Costa1
1Serviço de Coloproctologia do Hospital Geral de Goiânia - Goiânia (GO), e Instituto de Coloproctologia de Goiânia - Goiânia (GO) - Brasil.
RESUMO: Objetivo: estudar o tratamento e a evolução de 7 casos de tumor carcinóide de reto. Pacientes e método: análise retrospectiva do prontuário de 7 pacientes atendidos no Hospital Geral de Goiânia e Instituto de Coloproctologia de Goiânia. Resultados: 7 casos de tumor carcinóide de reto foram diagnosticados incidentalmente durante colonoscopias realizadas por indicações diversas. Em todos os casos foram realizadas polipectomias endoscópicas. Análises histológicas e imunohistoquímicas evidenciaram tumor carcinóide em todos os casos. Realizada retossigmoidectomia anterior em dois casos, devido a comprometimento da camada muscular da mucosa e excisão local transanal em um caso, devido a evidência de neoplasia atípica. O seguimento médio foi de 28 meses com evolução satisfatória em todos os casos. Conclusão: Os 7 pacientes com tumor carcinóide de reto estudados foram inicialmente tratados com ressecção endoscópica, tendo evolução satisfatória e mantendo-se livres de doença no período de seguimento.
Descritores: Tumor carcinóide de reto, tumor neuroendócrino
Introdução
Os tumores carcinóides foram descritos
pela primeira vez em 1888, por Lubarsch, que
encontrou múltiplos tumores no íleo distal de 2 pacientes
em autópsias. O termo Karzinoide foi usado
por Oberndorfer em 1907 para descrever tumores
ileais que tinham um comportamento mais benigno do
que os adenocarcinomas típicos 3, 12, 17, 21, 24,26, 27.
Fazem parte dos chamados tumores neuroendócrinos e constituem neoplasias
raras2, 6, 22, 25.originadas de um precursor celular comum, as
células neuroendócrinas que no trato digestivo são
chamadas células de Kulchitsky e originam-se das criptas
de Lieberkün2, 4, 12, 19, 20, 21, 24, 26.
Essas células são caracterizadas
histologicamente por reações positivas a sais de prata e
a marcadores de tecido neuroendócrino
incluindo enolase, sinaptofisina e
cromogranina.3, 26.
Têm a capacidade de produzir, armazenar e secretar diversos tipos de
neuropeptídeos biologicamente funcionantes ou
não-funcionantes2, 12, 20, 25, sendo a serotonina a substância principal e
também a mais estudada3, 11, 25. Essa substância é sintetizada
de seu precursor, 5-hidroxitriptofano, pela enzima aromática decarboxilase ácida, que é
subseqüentemente metabolizado pela monoamino-oxidase em ácido
5-hidroxiindolacético (5-HIAA), o qual é secretado
na urina2, 3 e pode auxiliar no diagnóstico de
tumores funcionantes 11. Essas células produzem
outras substâncias, como corticotropina, histamina,
dopamina, substância P, neurotensina, prostaglandina,
calicreína, entre outras3.
A Síndrome Carcinóide ocorre quando
a serotonina é liberada na circulação sistêmica,
causando manifestações clínicas de rubor,
diarréia, broncoespasmo e eventualmente, comprometimento
do coração direito2, 3, 14, 20, 21, 25
quando persiste por tempo prolongado. Essa síndrome está associada a
metástase hepática, na maior parte das
vezes2, 25.
Estima-se que até 50% dos pacientes com tumores carcinóides já tenham metástase
ao diagnóstico15.
São tumores de crescimento lento2, 7, 15, 16, 19,
23, 25, o que muitas vezes dificulta o diagnóstico em
fases iniciais12, 20.
Foram classificados em 1963, por Williams e Sandler, de acordo com seu sítio embriológico
de origem em Foregut (timo, pulmão, estômago,
pâncreas, vesícula biliar e duodeno), Midgut (intestino
delgado, apêndice e cólon direito), e Hindgut (cólon esquerdo
e reto)1, 2, 3, 5, 12, 14. Carcinóides originados de
diferentes sítios não são apenas histologicamente
e funcionalmente diferentes, mas também
possuem comportamentos diferentes17.
Uma classificação mais recente,
padronizada pelo WHO (World Health Organization), leva
em consideração o tamanho tumoral, a invasão na
parede e a presença ou não de metástases. Consiste em
quatro tipos: neoplasia endócrina bem diferenciada
de comportamento benigno, neoplasia endócrina
bem diferenciada de comportamento incerto,
carcinoma endócrino bem diferenciado (baixo grau
de malignização) e carcinoma endócrino
pouco diferenciado (alto grau de
malignização)1, 2, 5.
Até o momento, não há classificação
TNM para esses tumores1.
Os tumores carcinóides estão associados a
uma incidência aumentada de outros tumores
malignos, especialmente do trato gastrointestinal,
independentemente do seu sítio de origem. Uma
possível razão para o aumento na incidência de outros
cânceres poderiam ser as propriedades tumorigênicas
dos peptídeos secretados pelas células
neuroendócrinas24.
Os carcinóides de reto foram descritos
pela primeira vez em 1912 e correspondem a 1-2% de
todos os tumores de reto1, 2, 20. Em contraste com
os carcinóides de intestino delgado e cólon, que
produzem mais serotonina, produzem principalmente glucagon
e peptídeos de glicetina2, 3. Caracteristicamente, a
grande maioria é
não-funcionante20.
Cinqüenta por cento dos tumores
carcinóides de reto são assintomáticos e
diagnosticados incidentalmente, por endoscopias feitas por
indicações diversas10, 20, 21,
24 e a síndrome carcinóide é rara,
apesar de potencialmente metastatizante8,
20.
Aproximadamente 10% dos carcinóides
retais podem estar associados a outra neoplasia
colônica2. O tumor carcinóide apresenta-se como lesão
polipóide, séssil, endurecida, móvel, de localização submucosa
e de aspecto amarelado, ao exame macroscópico, o
que denota alto teor lipídico20, 21,
24 (Figura -1). O potencial de malignização e o prognóstico dos
tumores carcinóides de reto têm sido intimamente
relacionados com o seu tamanho2. As metástases nesses
tumores ocorrem principalmente para fígado e
linfonodos regionais10, 18.
|
|
| Figura 1 - Tumor carcinóide de reto |
A invasão ou não da muscular da
mucosa determina se a excisão endoscópica é suficiente ou
se é necessária complementação cirúrgica nos casos
de carcinóides retais ressecados
endoscopicamente. Recentemente, aceita-se que lesões menores que
1cm de diâmetro podem ser adequadamente tratadas
por ressecção endoscópica se margens negativas
após avaliação histológica forem
obtidas10, 12, 21. Outra abordagem seria a excisão local transanal, uma vez
que 75% dos tumores retais se localizam nos terços
médio e inferior do reto1, 3, 20
.
O tratamento de pacientes com tumores maiores que 2cm de diâmetro consiste na
ressecção abdominal anterior ou amputação abdominal de
reto1, 20. Entretanto, o valor desses procedimentos
no tratamento de carcinóides retais tem sido
recentemente questionado, uma vez que parece não aumentar
a sobrevida com relação àquela observada com a
excisão local, em estudos restrospectivos
3, 24.
A conduta em tumores de 1-2cm de diâmetro é controversa. Alguns autores sugerem que
pacientes com ulceração tumoral e invasão da muscular
da mucosa devem ser submetidos à cirurgia radical,
visto que esses fatores seriam de pior
prognóstico3, 20.
A taxa de sobrevida em 05 anos é de 81%
para pacientes com doença localizada, 47% para
pacientes com doença regional e 18% para pacientes
com metástase à
distância2.
OBJETIVO
Avaliar o tratamento e a evolução de 07
casos de tumor carcinóide de reto de até 2cm de
diâmetro, diagnosticados no Instituto de Coloproctologia
de Goiânia e Hospital Geral de Goiânia.
PACIENTES E MÉTODO
Análise retrospectiva do prontuário de
07 pacientes acompanhados no Instituto de Coloproctologia de Goiânia e no Hospital Geral de
Goiânia, no período de abril de 1998 a abril de 2005.
RESULTADOS
Foram estudados 3 homens e 4 mulheres com idade média de 53 anos (33-70). Quadro-1.
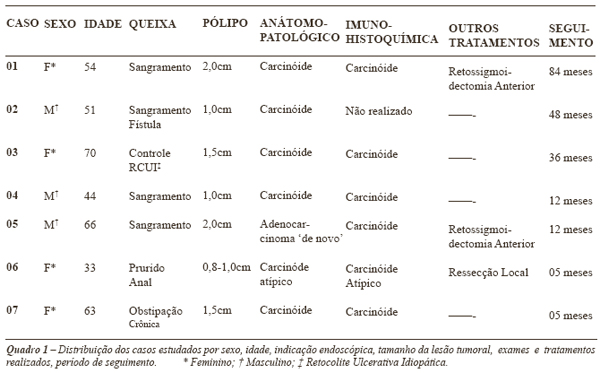 |
A maioria dos pacientes procurou o
serviço com queixas decorrentes de doença colorretal
orificial e o diagnóstico de tumor carcinóide foi
feito incidentalmente através de colonoscopia por
causas diversas. Uma paciente era portadora de
retocolite ulcerativa.
O tamanho tumoral variou de 0,8 a 2,0cm de diâmetro e em todos os casos foi feita
ressecção endoscópica.
O exame anátomo-patológico das
lesões diagnosticou tumor carcinóide de reto em todos
os casos, com exceção de um em que houve dúvida
entre adenocarcinoma "de novo" ou tumor
neuroendócrino. Em um dos casos estudados, o da paciente mais
jovem, o exame anátomo-patológico diagnosticou
tumor carcinóide atípico.
Em 6 dos 7 casos foi realizado estudo imunohistoquímico que confirmou o diagnóstico
de tumor carcinóide de reto.
Em dois pacientes as lesões apresentavam 2,0cm de diâmetro e a histologia mostrou invasão
da muscular da mucosa. Ambos foram submetidos a retossigmoidectomia anterior para complementação
do tratamento. A avaliação histológica das peças
cirúrgicas não mostrou doença residual.
A paciente mais jovem cujo diagnóstico
foi tumor carcinóide atípico, foi submetida a
ressecção local transanal, além da polipectomia, e o
exame anátomo-patológico da peça cirúrgica também
não mostrou doença residual.
Todos os pacientes foram seguidos com exame colonoscópico de controle, com freqüência
variável de acordo com o cirurgião. Esses exames
foram normais durante o período de seguimento, com
exceção de uma paciente, que havia sido submetida
a retossigmoidectomia anterior cujo exame mostrou pólipo em cólon transverso e o exame
anátomo-patológico da lesão revelou pólipo hiperplásico.
Todos os pacientes permaneceram sem
doença no período de seguimento, que foi, em média, de
28 meses.
DISCUSSÃO
A distribuição por sexos e por faixa etária
em nosso estudo está de acordo com a literatura que
mostra predomínio dos tumores neuroendócrinos no
sexo feminino4, 14, 17 e faixa etária mais atingida em torno
da sexta década de vida3; dentre nossos pacientes, 4
eram mulheres e 3 eram homens, e a média de idade foi
de 53 anos.
Todos os pacientes estudados eram assintomáticos e foram diagnosticados incidentalmente,
por exames colonoscópicos solicitados devido a
outras patologias. Na literatura, estima-se que o
diagnóstico incidental ocorra em torno de 50% dos casos
para carcinóides de reto10, 20. Os pacientes que têm
sintomas geralmente apresentam sangramento retal, dor,
ou constipação8. Uma de nossas pacientes
apresentava manifestações de retocolite ulcerativa. Há relatos
de uma associação aumentada entre o tumor
carcinóide de reto e essa doença
inflamatória24.
Todos os pacientes foram submetidos a polipectomia e a exame histopatológico, que é
o determinante do diagnóstico (Figuras-2 e
3). Atualmente faz-se confirmação por
estudo imunohistoquímico, através da pesquisa
dos marcadores tumorais de células
neuroendócrinas, entre eles a cromogranina A e a
sinaptofisina1, 2, 5, 11, 13, 25. Neste estudo, apenas um caso não foi
submetido ao estudo imunohistoquímico, devido à amostra
ser insuficiente.
 |
| Figura 2 - Tumor carcinóide de reto |
 |
| Figura 3 - Polipectomia endoscópica |
Nos dois casos em que os diâmetros
tumorais foram de 2,0cm, houve comprometimento de
muscular da mucosa e os pacientes necessitaram de
tratamento adicional com retossigmoidectomia, uma vez que
há íntima relação entre o tamanho tumoral e a
presença de metástases2, 10,
18. Elas ocorrem em aproximadamente 2% dos pacientes com lesões menores que 1cm
de diâmetro, enquanto lesões entre 1-2cm
apresentam metástases em 10-15% dos casos. Pacientes
com tumores maiores que 2cm freqüentemente
apresentam metástases em 60-80% dos casos
2, 3, 20.
Uma das pacientes estudadas apresentou carcinóide atípico e foi submetida a excisão local.
Há controvérsias na literatura sobre o conceito de
tumor "atípico", mas é consenso que este possui mais de
2 mitoses/mm2 e mais de 2% de células Ki-67+
(status proliferativo), sugerindo um comportamento
mais agressivo nesse tipo tumoral20, em contraste com
a maioria dos tumores neuroendócrinos, que
possuem monomorfismo celular com ausência ou baixo grau
de atipia (< 2 mitoses/ mm2) e baixo status
proliferativo (< 2% de células Ki-67 +), indicando um
crescimento lento e comportamento benigno1, 2,
26. Essa paciente não necessitou de procedimento adicional, uma vez que
não apresentou invasão de muscular da mucosa ao
exame histológico.
CONCLUSÃO
Os 7 pacientes do presente estudo que, submetidos a colonoscopia por indicações
diversas, tiveram diagnóstico incidental de tumor carcinóide
de reto menores que 2 cm de diâmetro, foram
todos primeiramente tratados com ressecção endoscópica,
em concordância com a literatura, tendo
evolução satisfatória e mantendo-se livres da doença no
período de seguimento.
SUMMARY: Objective: The aim of the present study was to analyze the treatment and follow-up of seven patients who suffered from rectal carcinoid tumor. Patients and Method: The authors have reviewed the records of seven patients operated on at the Hospital Geral de Goiânia and at the Instituto de Coloproctologia de Goiânia. Results: Seven cases of rectal carcinoid tumors were accidentally found during full colonoscopy assesment due to varied complaints. All of them were treated with endoscopic polipectomy and microscopy and immunochemistry have confirmed carcinoid tumor. Two patients underwent anterior resection due to invasion of the muscularis mucosae and one patient underwent transanal local excision due to atypical changes. The average follow-up time was 28 months and all the patients had an uneventful postoperative course.
Key words: Rectal carcinoid tumor, Neuroendocrine tumor.
Referências Biliográficas
1. Ramage JK, Davies AHG, Ardill J, Bax N, Caplin
M, Grossman A. et al. Guidelines for the management
of gastroenteropancreatic neuroendocrine (including
carcinoid) tumours. Gut. 2005; 54 (IV):1-16.
2. Kaltsas GA, Besser GM, Grossman AB. The diagnosis
and medical management of advanced neuroendocrine
tumors. Endocrine Reviews 2004; 25(3): 458-511.
3. Kulke M, Mayer RJ. Medical Progress: Carcinoid Tumors.
N Engl J Med. 1999; 340 (11): 858-868.
4. Maggard MA, O'Connell JB, Ko CY.Update
population-based review of carcinoid tumors. Ann Surg. 2004;
240(1): 117-122.
5. Öberg K. Carcinoid Tumors: molecular genetics,
tumor biology, and update of diagnosis and treatment. Curr
Opin Oncol.2002; 14(1): 38-45.
6. Öberg K. Neuroendocrine tumors of the gastrointestinal
tract: recent advances in molecular genetics, diagnosis,
and treatment. Curr Opin Oncol.2005; 17(4): 386-391.
7. Pelley RJ, Bukowski RM. Recent advances in systemic
therapy for gastrointestinal neuroendocrine tumors. Curr
Opin Oncol.1999; 11(1):32.
8. Maroy B. Similar Rectal Carcinoid Tumors of two
sibilings with curative endoscopic snare resection. J Clin
Gastroenterol. 1997; 24(2): 124-125.
9. Sharma R, McLeod D, Clarke SJ. Small blue cell tumors
of the rectum: Case 3. Atypical carcinoid of the rectum. J
Clin Oncol. 2005; 23(4): 914-915.
10. Banzo J, Vidal-Sicat S, Prats E, Galofré G, Razola P, Mañé
S et al. In-111 DTPA octreotide scintigraphy and
intraoperative gamma probe detection in the diagnosis and treatment
of residual lymph node metastases of a rectal carcinoid
tumor. Clin Nucl Med. 2005; 30(5) 308-311.
11. Taupenot L, Harper KL, O'Connor DT. Mechanisms
of disease: the chromogranin-secretogranin family. N Engl J
Med. 2003; 348(12): 1134-1149.
12. Shebani KO, Souba WW, Finkelstein DM, Stark PC,
Elgadi KM, Tanabe KK et al. Prognosis and survival in patients
with gastrointestinal tract carcinoids tumors.
Ann Surg. 1999; 229(6): 815.
13. Erickson LA, Lloyd RV. Practical markers used in the
diagnosis of endocrine tumors. Adv Anat Pathol 2004. 11(4): 175-189.
14. Dierdorf SF. Carcinoid tumor and carcinoid syndrome.
Curr Opin Anaesthesiol 2003; 16(3): 343-347.
15. Jensen RT. Carcinoid and pancreatic endocrine tumors:
recent advances in molecular pathogenenesis, localization
and treatment. Curr Opin Oncol 2000; 12(4): 368-377.
16. Cheng JY, Sheu LF, Meng CL, Lin JC. Expression of p53
in colorectal carcinoids. Arch Surg. 1996; 131(1): 67-70.
17. Onaits MW, Kirshbom PM, Hayard TZ, Quayle FJ,
Feldman JM, Seigler HF et al. Gastrointestinal
carcinoids: characterization by site of origin and hormone
production. Ann Surg. 2000; 232(4): 549-556.
18. Venook AP. Embolization and chemoembolization therapy
for neuroendocrine tumors. Curr Opin Oncol. 1999; 11(1): 38.
19. Barakat MT, Meeran K. Neuroendocrine tumors and
gut hormones. Curr Opin Endocrinol Diabet 2005; 12(1): 99-105.
20. Rossi BM, Nakagawa WT, Ferreira FO, Aguiar Jr S, Lopes
A . Câncer de Cólon, Reto e Ânus. Ed Marina e Ed Tecmedd. 2005; 40: 535-538.
21. Maizier WP, Levien DH, Luchtefeld MA, Senagore
AJ. Surgery of the Colon, Rectum, and Anus. W. B.
Saunders Company. 1995; 47: 544-545.
22. Boudreax JP, Putty B, Frey DJ, Woltering E, Anthony L,
Dally I et al. Surgical treatment of advanced-stage carcinoid tumors:
lessons learned. Ann Surg. 2005; 241(6): 839-846.
23. Kwekkeboom DJ, Teunissen JJ, Bakker WH, Kooij PP,
de Heder WW, Feelders RA et al. Radiolabeled
somatostatin analog [177Lu-DOTAo, Tyr3] octreotate in patients
with endocrine gastroenteropancreatic tumors. J
Clin Oncol.2005.23(12): 2754-2762.
24. Corman ML: Colon & Rectal Surgery. Philadelphia.5a.
Ed. Lippincott Williams & Wilkins, 2005.p.1091-1099.
25. Lal A, Chen H. Treatment of advanced carcinoid tumors.
Curr Opin Oncol. 2006; 18(1): 9-15.
26. Fenwick SW, Wiatt JI, Toogood GJ, Lodge JPA.
Hepatic resection and transplantation for primary carcinoid tumors of
the liver. Ann Surg. 2004; 239 (2): 210-219.
27. Yantiss RK, Odze RD, Farraye F, Rosenberg AE.
Solitary versus multiple carcinoid tumors of the ileum: a clinical
and pathologic review of 68 cases. Am J Pathol. 2003; 27(6):
811-817.
Endereço para correspondência:
Patrícia Alves Mangueira
Av: C n. 1129 - Quadra 41 - Lote 38 - Setor Couto Magalhães
77.824-780 - Araguaina (TO)
Recebido em 04/04/2006
Aceito para publicação em 08/05/2006
Trabalho realizado no Serviço de Coloproctologia do Hospital Geral de Goiânia _ Goiânia (GO) e no Instituto de Coloproctologia de Goiânia - Goiânia (GO) - Brasil.