
ARTIGOS ORIGINAIS
POLIPOSE ADENOMATOSA FAMILIAR ATENUADA
Attenuated Familial Adenomatous Polyposis
Gabriella Oliveira Fernandes1; Jaime JosÉ Pereira Junior1; Marco AurÉlio Viana FranÇa1; JosÉ Hermes Gomes Costa1
1Hospital Geral de Goiânia. Goiânia - GO - Brasil.
RESUMO: A Polipose Adenomatosa Familiar Atenuada(PAFA) é uma síndrome autossômica dominante, de diagnóstico tardio, comparando-se à forma clássica da polipose adenomatosa familiar. Dentre as características da síndrome estão: a)presença de menos de 100 pólipos colorretais; b) curso brando da doença, com idade tardia do diagnóstico e do aparecimento de câncer; c)prevalência maior dos pólipos à direita do cólon; d) reto poupado de lesões, na maioria dos casos. Analisar as características clínicas, tratamento e seguimento de 13 pacientes com diagnóstico de PAFA. Dos pacientes estudados, a média de idade ao diagnóstico foi 55 anos. Cinco pacientes apresentavam história familiar de polipose e/ou neoplasia. Nove (69%) pacientes já tinham câncer no momento do diagnóstico. A maioria dos pacientes possuía pólipos localizados no cólon direito (31%). Do total, 06 pacientes foram submetidos à ressecção cirúrgica, com proctocolectomia ou colectomia. A média de seguimento dos pacientes foi de 26 meses. O controle foi realizado através de colonoscopias e retossigmoidoscopias, de acordo com o tratamento realizado. O diagnóstico de PAFA foi feito em idade tardia em relação à forma clássica da doença, com a maioria dos pólipos localizados no cólon direito. O controle endoscópico dos pacientes deve ser realizado com rigor. A colectomia com anastomose do íleo-reto é uma boa opção cirúrgica no tratamento dos pacientes, com baixa recidiva de pólipos no reto.
Descritores: Polipose familiar, polipos colonicos, adenoma.
INTRODUÇÃO
A Polipose Adenomatosa Familiar Atenuada
(PAFA) constitui síndrome autossômica
dominante, relacionada a mutações no gene APC, porém
com características distintas da Polipose
Adenomatosa Familiar(PAF): a) curso brando da doença, com
idade tardia no diagnóstico e no aparecimento de
câncer colorretal (10-15 anos mais tarde que na
PAF); b)presença de menos de 100 pólipos colorretais,
na maioria dos casos; c) maior distribuição proximal
dos pólipos e neoplasias no cólon; d) reto poupado das
lesões, na maioria dos
casos1,2,5,6,12,13.
São descritas lesões neoplásicas
sincrônicas nesses pacientes e sabe-se que o reto é poupado
da presença de pólipos em grande parte dos
pacientes. Por ser incomum, a incidência e freqüência da
PAFA não são conhecidas, embora se estime que cerca
de 10% dos pacientes apresentem o fenótipo atenuado
da síndrome1, 2.
O diagnóstico da doença é baseado nos
achados clínicos e endoscópicos, além de testes
genéticos como o Teste da Proteína Truncada e o
mapeamento de DNA1,2,4,6,7,13. Pacientes com endoscopia
digestiva alta (EDA) evidenciando pólipos gastro-duodenais,
ou história familiar de polipose, e/ou neoplasia
colorretal devem ser investigados com colonoscopia
para PAFA1,13. Assim como na polipose clássica,
manifestações extra-colônicas podem estar presentes em
menor escala, como tumores desmóides e hipertrofia
congênita do epitélio pigmentar da
retina1,2. Os pólipos gátricos e duodenais são as manifestações mais
comuns, com relatos de transformação maligna dos
mesmos8,9.
O gene APC é um supressor tumoral,
agindo como barreira na seqüência adenoma-carcinoma.
Dentre suas funções está a regulação intracelular de
b-catenina, uma proteína associada ao fator de
transcrição de células T, relacionada à transcrição de DNA
e envolvida em processos de sinalização celular que
afetam a apoptose e o crescimento das
células1,2,13. Mutações no APC levam à perda de sua função
reguladora, com aumento de b-catenina, proliferação
celular, além da transcrição de genes envolvidos no
crescimento do epitélio colônico, incluindo o oncogene
c-myc2,6,7. Portanto, há uma redução e perda da
atividade supressora tumoral. Hoje, são descritas cerca de
34 mutações no gene APC relacionadas à PAFA,
detectadas em 3 partes do gene : extremidade 5' (5
exons iniciais), exon 9 e extremidade distal 3'
1,2,12,13. Apesar de tais descobertas, cerca de 20% a 50% dos
pacientes com PAF não apresentam qualquer mutação
no gene APC, levando à hipótese de que genes ainda
desconhecidos, fatores de crescimento, hormonais e ambientais estejam associados à fisiopatogenia da
doença, resultando em características fenotípicas
atenuadas 2,6,12,13.
Estudos têm mostrado a presença de
vários fenótipos em pacientes portadores de uma
mesma mutação, com manifestações colônicas e
extra-intestinais distintas, além de diferenças na progressão
da doença 1,2,6 A grande variedade fenotípica
apresentada pelos pacientes dificulta a padronização de
critérios diagnósticos para PAFA, além de casos
confundidos com HNPCC e câncer colorretal (CCR)
esporádico4,5,13. Não se sabe ainda o verdadeiro risco de
CCR na PAFA, sendo a indicação de colectomia
profilática ainda controversa na
literatura3,13. Sabe-se que em pacientes com poucos pólipos o controle endoscópico
pode ser realizado periodicamente, além do uso de
derivados inibidores da COX-2 (Celecoxib) na prevenção
e redução dos
pólipos5,10,11.
Nos casos cujo controle endoscópico seja
difícil, ou ainda considerando-se o potencial maligno
dos pólipos, indica-se a colectomia total com
anastomose do íleo-reto, ou proctocolectomia com bolsa
íleo-anal3. Pacientes, cuja cirurgia preservou o reto, devem
ser monitorizados com exames periódicos. Nos
pacientes não operados, o seguimento é preferível através
de colonoscopia, devido à localização das lesões ser
mais comum no cólon direito
1,12,13. O controle pode ser feito em períodos anuais, podendo-se utilizar técnicas
de coloração que facilitem a identificação dos pólipos.
OBJETIVO
Analisar as características demográficas,
manifestações colônicas e extra-colônicas,
tratamento realizado e evolução clínica de 13 pacientes com
PAFA, atendidos no Hospital Geral de Goiânia no período
de março de 1995 a junho de 2006.
PACIENTES E MÉTODOS
Estudo retrospectivo através da análise de
prontuários de 13 pacientes atendidos na referida
instituição com diagnóstico clínico de PAFA. Os critérios
de inclusão foram: a) presença de menos de 100
pólipos colônicos ao exame endoscópico; b) idade de
diagnóstico dos pólipos e / ou câncer colorretal superior a
35 anos; c) exclusão de critérios para HNPCC.
Foram analisados dados clínicos, endoscópicos,
tratamento realizado e a evolução dos pacientes. Testes
genéticos não foram realizados, devido à indisponibilidade
dos mesmos.
RESULTADOS
Quanto ao sexo, 08(62%) eram do sexo feminino e 5 (38%) do sexo masculino (gráfico 1). A média
de idade no momento do diagnóstico foi de 55 (37-69)
anos (Tabela 1). Cinco pacientes possuíam história familiar
de polipose e/ou neoplasia colorretal; dentre eles, 04
(80%) já apresentavam CCR no momento do diagnóstico.
Os principais sintomas relatados foram: dor abdominal, sangramento retal, alteração do ritmo
intestinal, anemia, nodulação anal e perda ponderal
(gráfico 2). A localização dos pólipos ao
exame colonoscópico foi a seguinte, em frequência: 02
pacientes no cólon esquerdo (15,38%), 04 pacientes no
cólon direito(30,77%); 04 pacientes em todo o
cólon(30,77%) e 03 pacientes em todo o cólon e reto (23,08%)
(gráfico 3)(Foto1). O exame histopatológico dos
pólipos mostrou adenomas tubulares, adenomas vilosos
e túbulo-vilosos, inclusive no mesmo paciente, com
graus de atipia variados, além de pólipos hiperplásicos.
Dos 13 pacientes avaliados, 09(69%) apresentavam câncer no momento do diagnóstico, assim
distribuídos: 06(66,7%) no cólon direito; 02 (22,2%) no
reto; e 01(11,1%) no cólon esquerdo (Foto2).
|
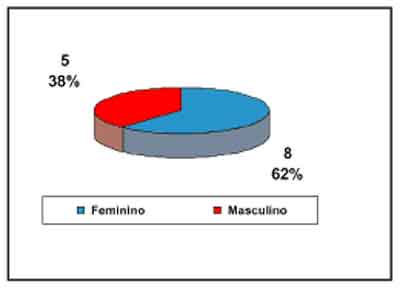 |
| Gráfico 1 - Distribuição quanto ao sexo dos pacientes. |
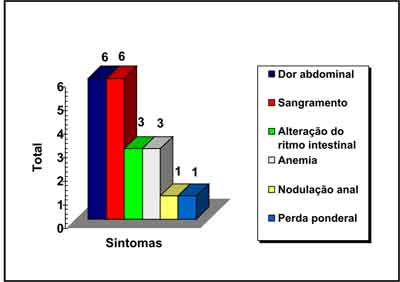 |
| Gráfico 2 - Sintomas dos pacientes. |
 |
| Gráfico 3 - Localização dos pólipos. |
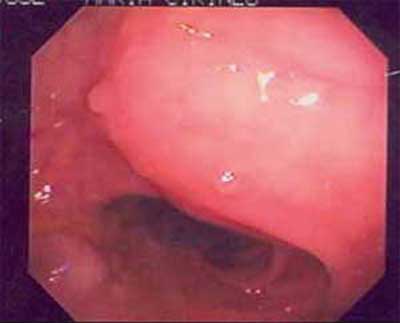 |
| Figura 1 - Colonoscopia com pólipos no cólon direito. |
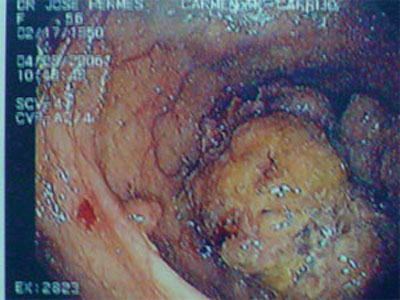 |
| Figura 2 - Colonoscopia mostrando pólipose associada a tumor de cólon direito. |
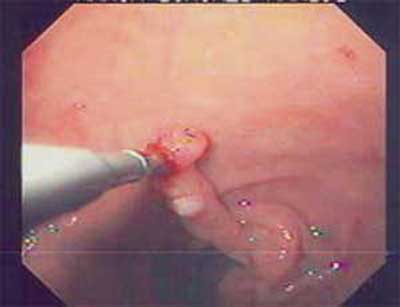 |
| Figura 3 - EDA com presença de pólipo duodenal. |
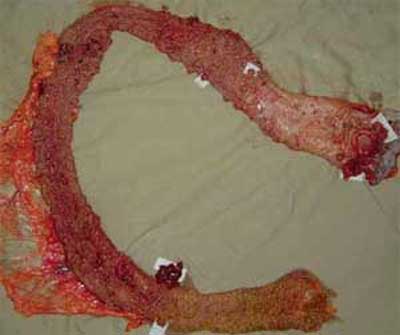 |
| Figura 4 - Peça cirúrgica de proctocolectomia, com tumoração em reto e polipose. |
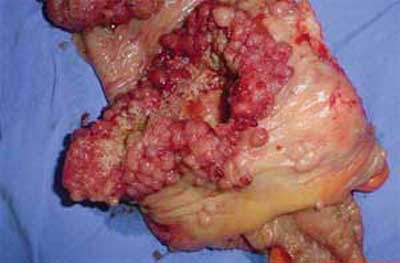 |
Figura 5 - Peça cirúrgica com tumoração em ceco. |
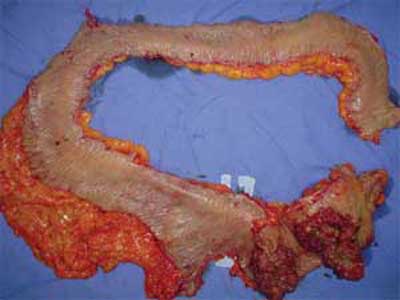 |
| Figura 6 - Peça cirúrgica de colectomia total, com tumor sincrônico de ceco e cólon ascendente. |
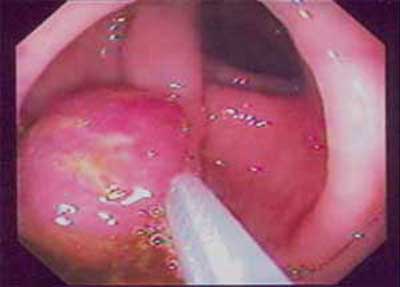 |
| Figura 7 - Polipectomia durante controle pós-operatório em colonoscopia. |
DISCUSSÃO
Neste estudo retrospectivo, avaliamos as características de 13 pacientes portadores de
PAFA, doença autossômica dominante de diagnóstico
tardio, em relação à forma clássica da PAF. A média de
idade no momento do diagnóstico neste estudo foi de
55 anos, compatível com trabalhos de Soravia (58
anos)13 e Lamlum (54 anos)14. O diagnóstico foi baseado
nas características clínicas da doença já descritas,
além de história familiar de pólipos e/ou CCR. Alguns
autores têm referido pacientes com PAFA com
número de adenomas excedendo o critério de 100 lesões,
devido à forma branda da doença e diagnóstico
tardio da mesma15,16,17. A colonoscopia é o exame de
escolha para o diagnóstico e
screening nos pacientes, devido à localização comum dos pólipos à direita
do cólon, sendo o reto poupado na maioria dos
casos. Na nossa casuística, 31% dos pacientes
apresentavam a distribuição das lesões à direita do cólon, e
em 23% haviam pólipos no reto. Em série de
Soravia13, em 11 famílias com 79 membros com diagnóstico
de PAFA, grande parte deles apresentavam menos de 100 adenomas, com tendência a se localizarem à
direita do cólon, estando o reto livre de lesões em
todos os casos. A cirurgia está indicada nos pacientes
com CCR, não havendo consenso na literatura quanto
à colectomia profilática.
Embora a penetrância de CCR pareça
elevada, a incidência, freqüência e sobrevida dos
pacientes não são conhecidas. Neste estudo, 09 dos 13
pacientes já apresentavam câncer no momento
do diagnóstico, compatível com relatos de
Soravia13 (24/79) e Spirio16 (44/90).
Lynch18 preconiza colonoscopias de controle com polipectomias em pacientes com
poucas lesões, e segundo Hernegger et
al12, a colectomia está indicada naqueles com número elevado de
lesões, impossibilitados de seguimento endoscópico,
ou cujas polipectomias não sejam factíveis
tecnicamente. No nosso estudo houve apenas 01 óbito durante
o seguimento dos pacientes, devido a câncer
avançado com metástases.
Retossigmoidoscopia de controle, diferente da PAF, está indicada apenas nos pacientes com
anastomose do íleo-reto. Neste trabalho, dos 05 pacientes
submetidos a este exame no seguimento, 01 paciente
apresentou pólipo retal após 04 anos da cirurgia, confirmando
a necessidade de avaliação do reto preconizada por
Clark19 e Hernegger13, mesmo com a ausência freqüente
de pólipos neste segmento intestinal em casos descritos
na literatura. A endoscopia digestiva alta deve fazer
parte do controle desses pacientes, avaliando-se
manifestações da doença no trato gastrointestinal alto.
CONCLUSÃO
![]() O diagnóstico e a inclusão dos
pacientes com PAFA, neste estudo, foram baseados em
dados clínicos e endoscópicos, ocorrendo em idade tardia,
com câncer colorretal presente na maioria dos pacientes.
O diagnóstico e a inclusão dos
pacientes com PAFA, neste estudo, foram baseados em
dados clínicos e endoscópicos, ocorrendo em idade tardia,
com câncer colorretal presente na maioria dos pacientes.
![]() A sintomatologia levou à realização
de colonoscopia diagnóstica, com a maioria dos
pólipos localizados proximal à flexura esplênica.
A sintomatologia levou à realização
de colonoscopia diagnóstica, com a maioria dos
pólipos localizados proximal à flexura esplênica.
![]() Tumores sincrônicos ocorreram em 02
pacientes.
Tumores sincrônicos ocorreram em 02
pacientes.
![]() Colectomia total com anastomose do
íleo-reto é uma boa opção cirúrgica para os pacientes
com PAFA, com baixa recidiva dos pólipos no reto.
Colectomia total com anastomose do
íleo-reto é uma boa opção cirúrgica para os pacientes
com PAFA, com baixa recidiva dos pólipos no reto.
![]() É necessário o controle endoscópico
dos pacientes não operados, além de pesquisa de
outras manifestações extra-colônicas da doença.
É necessário o controle endoscópico
dos pacientes não operados, além de pesquisa de
outras manifestações extra-colônicas da doença.
![]() Familiares dos pacientes com
diagnóstico de PAFA devem ser orientados, para que se faça
o diagnóstico precoce da doença.
Familiares dos pacientes com
diagnóstico de PAFA devem ser orientados, para que se faça
o diagnóstico precoce da doença.
ABSTRACT: Attenuated Familial Adenomatous Polyposis (AFAP) is a heritable autosomally dominant syndrome, with
later diagnosis than the classical condition of Familial Adenomatous Polyposis. Amid its main features there are : a) the presence
of less than 100 polyps; b) the mild course of the disease and its later diagnosis and development of colon cancer; c)the polyps
are more frequent in the right colon; d)the rectum may be relatively or even totally spared. To analyze the clinical
manifestations, treatment and follow-up of 13 patients with AFAP. The mean age was 55 years, five patients had positive family history
of polyposis and/or colon cancer and nine (69%) patients had already developed colonic cancer at the time of the diagnosis. Most
of the patients had polyps located in the right colon. Six out of 13 patients patients had undergone surgical resection,
either proctocolectomy or colectomy. The average follow-up time was 26 months. Periodically colonoscopy or retosigmoidoscopy
were employed for follow-up evaluation, according to the previous surgical procedure. The diagnosis of AFAP was made later than
the one of the classic form of the disease and most of the polyps were located in the right colon. Frequent follow-up with
endoscopic examination as a follow-up is mandatory. Colectomy with ileo-rectal anastomosis is a very good option in the surgical
management of these patients with low recurrence rate of rectal polyps.
Key words: familial polyposis, colonic polyps, adnome.
Referências
1. Hernegger GS, Moore HG, Guilem JG. Attenuated
Familial Adenomatous Polyposis: An evolving and poorly
understood entity.Dis Colon Rectum 2002; 45:127-136
2. Knudsen AL, Bisgaard ML, Bülow S. Attenuated
Familial Adenomatous Polyposis (AFAP).A review of the
literature. Familial Cancer 2003; 2:43-55.
3. Contessini-Avesani E, Botti F, Negri C
et al. Familial adenomatous polyposis. Surgical treatment: when and
how. Tech Coloproctol 2004; 8:S309-S314.
4. Half EE, Bresalier RS. Clinical management of
hereditary colorectal cancer syndromes.Curr Opin
Gastroenterol 2004;20:32-42.
5. Esaki M, Matsumoto T, Mizuno M et
al. Effect of Sulindac treatment for attenuated familial adenomatous polyposis
with a new germline APC mutation at codon 161. Dis Colon
Rectum 2002;45:1397-1406.
6. Moision A-L, Järvinen H,
Peltomäki.Genetic and clinical characterisation of familial adenomatous polyposis:
a population based study.Gut 2002;50:845-850.
7. Neklason DW, Solomon CH, Dalton AL et
al. Intron 4 mutation in APC gene results in splice and attenuated
FAP phenotype.Familial Cancer 2004;3:35-40.
8. Chetty R, Salahsho S, Bapat B et
al. Intraductal papillary mucinous neoplasm of the pancreas in a patient with
attenuated familial adenomatous polyposis. J Clin Pathol
2005;58:97-101.
9. Trimbath JD,Griffin C, Romans K et
al. Attenuated familial adenomatous polyposis presenting as
ampullary adenocarcinoma. Gut 2003;52:903-904.
10. Philips RKS, Wallace MH, Lynch PM et
al.A randomised, double blind, placebo controlled study of celecoxib, a
selective cyclooxygenase 2 inhibito, on duodenal polyposis in
familial adenomatous polyposis.Gut 2002; 50:857-860.
11. Peek Jr RM. Prevention of colorectal cancer through the
use of COX-2 selective inhibitors. Cancer Chemother
Pharmacol 2004;54(Suppl 1):S50-S56.
12. Su L-K, Kohlmann W, Ward PA. Different
familial adenomatous polyposis phenotypes resulting from
deletions of the entire APC exon 15. Hum Genet 2002; 111:88-95.
13. Soravia C, Berk T, Madlensky L et
al. Genotype-Phenotype correlations in attenuated adenomatous poliposis coli.Am
J Hum Genet 1998;62:1290-1301.
14. Lamlum H, Al Tassan N, Jaeger E. Germline APC variants
in patients with multiple colorectal adenomas, with evidence
for the particular importance of E1317Q. Hum Mol
Gen 2000;9:2215-2221.
15. The unique phenotype of the attenuated adenomatous
polyposis coli syndrome. Poster/LCPG-meeting 2001; Venice, Italy.
16. Spirio L, Green J, Robertson J et
al. The identical 5´splice-site acceptor mutation in five attenuated APC families
from Newfounland demonstrates a founder effect. Hum
Genet 1999;105:388-398.
17. van der Luijt RB, Vasen HF, Tops CM
et al. APC mutation in the alternatively spliced region of exon 9 associates with
late onset familial adenomatous polyposis coli patients.
Hum Genet 1995;96:705-710.
18. Lynch HT, Watson P. AFAP: variety is the spice of life.
Gut 1998;43:451-452.
19. Clark SK, Middleton SB, Philips RK. It is really not
familial adenomatous polyposis. Dis Colon Rectum 2000;43:113.
Endereço para correspondência:
Gabriella Oliveira Fernandes
Endereço : Hospital Geral de Goiânia - H.G.G.
Serviço de Coloproctologia
Av: Anhanguera, No. 6379 - Setor Oeste
CEP : 74043-011 Goiânia - GO
E-mail : gabicir@ig.com.br
Recebido em 30/11/2006
Aceito para publicação em 24/01/2007
Trabalho realizado no Serviço de Coloproctologia do Hospital Geral de Goiânia - H.G.G.