
ARTIGOS ORIGINAIS
POLIPOSE MÚLTIPLA FAMILIAR. ANÁLISE DE 44 CASOS TRATADOS NO HOSPITAL DAS CLÍNICAS DA FMRP-USP
Familiar Adenomatosis Polyposis: Analysis Of Forty-Four Cases from the School of Medicine of Riberão Preto Hospital and Clinics
Andreza Regina de B. M. da Silva¹; Rogério Serafim Parra¹; Jaicer GonÇalves Rolo²; Roberto Bueno Filho²; Omar FÉres³; JosÉ Joaquim Ribeiro da Rocha³
1 Pós-graduado da Área de Clínica Cirúrgica do Depto. de Cirurgia e Anatomia, FMRP-USP; 2 Aluno de graduação da FMRP-USP; 3 Professor Doutor da Disciplina de Coloproctologia, Depto. Cirurgia e Anatomia, FMRP-USP.
Resumo: Os autores apresentam análise retrospectiva de 44 pacientes com Polipose Múltipla Familiar tratados no Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto,Universidade de São Paulo, entre janeiro de 1991 a julho de 2005. Foram estudados aspectos epidemiológicos (idade, sexo), genéticos, principais sintomas, antecedentes pessoais e aspectos envolvendo tratamento cirúrgico e complicações pós-operatórias, comparando os achados com os da literatura correlata. Dos pacientes, 31 são do sexo masculino e 13 do feminino com idade média de 32 anos (14 - 60 anos). Os sintomas prevalentes foram: sangramento intestinal (62,5 %), alteração do hábito intestinal (60%), e com menor freqüência dor abdominal (45 %) e emagrecimento (30%). Relataram casos de polipose familiar 67,5% dos pacientes e 62,5% referiram parentes com antecedente de neoplasias (intestinal e extra-intestinal). Cerca de 32,5% dos pacientes já apresentavam neoplasia de cólon na época do diagnóstico da polipose,com idade média de 39 anos. O tratamento cirúrgico foi realizado em 95,4% dos pacientes: 35,7% foram submetidos à proctocolectomia total(9 casos com bolsa ileal em J e 6 casos com ileostomia definitiva) e 59,2% a colectomia total com ileorretoanastomose. Atualmente 57% dos pacientes avaliados ainda estão em seguimento com reavaliações periódicas, 7% faleceram e 27% abandonaram o tratamento.
Descritores: polipose familiar, câncer colorretal, colectomia, genética.
Introdução
A polipose adenomatosa familiar (PAF) é
uma doença hereditária , autossômica dominante
,com penetrância próxima de 100% , causada por uma
mutação no Adenomatous polyposis coli (APC) ,
localizado no cromossomo 5q 21. APC é um gene
supressor de tumor, identificado em 1987, e reproduzido em
1991 após análise de mutações em famílias com
PAF1.
Sua incidência é de aproximadamente 1
em 7000 a 1 em 16000 nascidos vivos , sendo
responsável por menos de 1% dos casos de câncer
colorretal2.
Mutações no APC são os passos iniciais
para o desenvolvimento do câncer colorretal. Ele está
relacionado com atividades celulares fundamentais nos
processos de produção de proteínas, adesão celular e
migração. Quando o APC sofre mutação, há
interferência na proliferação , apoptose e controle das
mudanças de fase do ciclo
celular3.
A identificação de alterações no gene APC
é feita com auxílio de testes genéticos e apesar disto,
20 a 30% dos pacientes com PAF não apresentam
alterações do gene no teste de rastreamento genético.
Nestes casos testes de maior sensibilidade, como o
MAMA (monoallelic mutation analysis) podem ser usados.
A combinação destes dois testes permite que em
95% dos casos as alterações do gene APC
sejam identificadas4.
A PAF se manifesta pela presença de numerosos pólipos adenomatosos em todo
trato gastrointestinal, principalmente o cólon. Estes
pólipos podem estar presentes vários anos antes do
aparecimento dos sintomas, fato que ratifica a
importâncias de investigação precoce dos familiares de
pacientes acometidos pela doença. Estudos têm afirmado a
existências de pólipos por pelo menos uma década
antes do aparecimento dos sintomas1. Estes pólipos
progridem para câncer colorretal até 35-40 anos se não
tratados adequadamente.
Os pólipos também acometem o trato
gastro-intestinal alto (estômago e duodeno) sendo que
aproximadamente 2/3 dos adenomas duodenais ocorrem
na região periampular5. Adenomas duodenais
avançados representam risco aumentado de câncer, sendo a
terceira causa de morte em pacientes com polipose adenomatosa familiar (8,2%), ficando atrás
das metástases do CCR (58,2%) e de tumores
desmóides (10,9%)6.
Outra forma de apresentação clínica é
a hipertrofia congênita do epitélio pigmentado da
retina (CHRPE), que se caracteriza pela presença de
lesões pigmentadas no exame de fundo de olho
ocorrendo em 70-80% dos pacientes com polipose.
Estas alterações oftalmológicas estão usualmente
presentes no nascimento, são assintomáticas, sem
potencial maligno e podem ser avaliadas por exames de
baixa complexidade7.
Tumores desmóides são mais raros,
ocorrendo em 15% dos casos de PAF8, mais freqüentemente
no abdome e parede abdominal (50%).
Preferencialmente são assintomáticos e lesões intraabdominais
podem causar dor ou podem ser complicadas por obstrução ureteral, hemorragias e até mesmo
fístulas enterocutâneas9.
O câncer da tireóide ocorre em 1 a 2%
dos casos. Tendem a ser bem delimitados, pouco
agressivos e com baixo potencial de
metástases10,11.
A associação de pólipos gastrointestinais
com osteomas caracteriza uma das variantes da PAF
, Síndrome de Gardner. Osteomas tipicamente
ocorrem na mandíbula e ossos longos. São tumores
benignos e indolores, e normalmente seu diagnóstico
precede o da polipose12.
A síndrome de Turcot é a associação da
PAF com tumores primários do Sistema Nervoso Central
, sendo mais comum o meduloblastoma13.
Polipose atenuada é uma variante
fenotípica distinta que se caracteriza pela presença de menos
de 100 pólipos colônicos e localizados
preferencialmente no cólon direito. Nestes casos o desenvolvimento
do câncer colorretal é mais tardio (15 anos mais tarde
do que nos casos clássicos)14.
Objetivo
O objetivo deste trabalho é estudar 44
pacientes com diagnóstico de polipose adenomatosa
familiar, analisando aspectos epidemiológicos, genéticos ,
principais sintomas ,antecedentes pessoais e o tipo de
tratamento proposto, avaliando também suas
complicações.
Material e métodos
Foi realizada avaliação retrospectiva do
prontuário de 44 pacientes acompanhados no Serviço
de Coloproctologia do HCRP-USP no período de
janeiro de 1991 a julho de 2005,obtendo-se os dados a
partir de um protocolo pré_elaborado. Os seguintes
dados foram revisados: prevalência de sexo e idade ,
principais sintomas apresentados, alterações no exame
físico, neoplasias associadas, antecedentes familiares
de polipose colônica e neoplasias. Também foi revisado
o tipo de tratamento cirúrgico e suas complicações
pós-operatórias, e fatores relacionados a qualidade de
vida pós-operatória como a presença de incontinência
fecal, necessidade do uso de medicamentos antidiarréicos
e o hábito intestinal após a cirurgia.
Após a coleta de dados, foi realizada a
análise da freqüência simples das variáveis
correlacionando os achados com os da literatura correlata.
Resultados
Foram avaliados 44 pacientes, sendo 31 do sexo masculino e 13 do feminino,com idade média de 32
anos (14-60 anos) (Tabela 1). Os sintomas prevalentes
foram sangramento intestinal (62,5%) , alteração do
hábito intestinal (60%), e com menor freqüência dor
abdominal (45%) e emagrecimento (30%).
|
 |
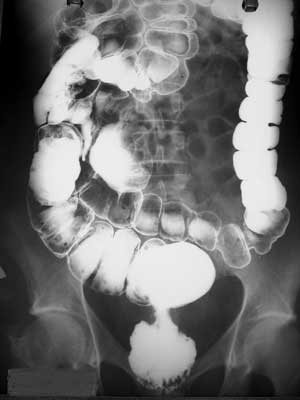 |
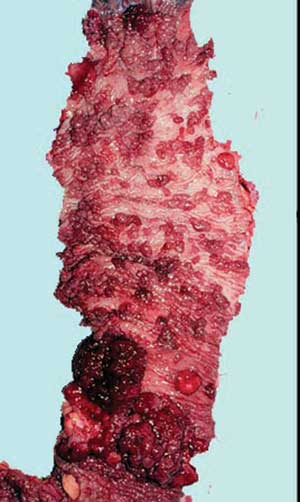
Figura 1A - Cólon acometido por polipose e com neoplasia. |
Figura 1B - Enema opaco evidenciando neoplasia estenosante
do reto. |
 |
 |
Figura 2 - Cólon acometido por polipose. |
 |
Figura 3 - Heredograma demonstrando indivíduos acometidos. |
 |
ABSTRACT: The authors present a retrospective analysis of forty-four patients with familiar adenomatosis polyposis treated at the School of Medicine of Ribeirão Preto Hospital and Clinics _ University of São Paulo, from January 1991 to July 2005. Epidemiologic (age and gender) and genetic aspects were investigated as well as main symptoms, personal history and surgical treatment outcome. Data obtained were compared to the available literature. Our results show that 31 patients were male and 13 female, with average age of 32 years-old (14 to 60 years-old). The main symptoms were intestinal bleeding (62.5 %) and intestinal habit alteration (60 %). Less frequently, patients showed abdominal pain (45 %) and loss of weight (30 %). Familiar adenomatosis polyposis was reported by 67.5 % of the patients and relatives with neoplasms (intestinal and extra-intestinal) were referred by 62.5 % of the patients. By the time of the polyposis diagnosis, 32.5 % of the patients have already been diagnosed for colon cancer, with average age of 39 years old. Surgical treatment was performed in 95.4 % of the patients, total proctocolectomy was performed in 35.7 % (9 cases with ileal J- pouch and 6 with permanent ileostomy) and total colectomy with ileo rectal anasthomosis in 59.2%. From all patients evaluated, 57 % are still under follow up, 7 % died and 27 % gave up treatment.
Key words: familiar polyposis, colorectal cancer, colectomy, genetics.
Referências
1. Groden J, Thliveris A, Samowitz W, et al. Identification
and characterization of the familial adenomatous polyposis
coli gene. Cell 1991; 66: 589-600.
2. Burt RW, Bishop DT, Lynch HT, et al. Risk and
surveillance of individuals with heritable factors for colorectal cancer.
WHO Collaborating Centre for the Prevention of Colorectal Cancer.
Bull World Health Organ 1990; 68: 655-65.
3. Näthke I. APC at a glance. J Cell Sci 2004;117: 4873-5
4. Laken SJ, Papadopoulos N, Petersen GM , et al. Analysis
of masked mutations in familial adenomatous polyposis.
Proc Natl Acad Sci USA 1999; 96: 2322-6.
5. Bertoni G, Sassateli R, Nigrisoli E, et al. High prevalence
of adenomas and microadenomas of the duodenal papilla
and periampulllary region in patients with familial
adenomatous polyposis. Eur J Gastroenterol Hepatol 1996; 8: 1201-6.
6. Arvantis ML , Jagelman DG,Fazio VW, et al. Mortality
in patients with familial adenomatous polyposis Dis
Colon Rectum 1990; 33: 639-42
7. Trabulsi El, Krush AJ, Gardner EJ, et al . Prevalence
and importance of pigmented ocular fundus lesions in
Gardner's syndrome. N Engl J Med 1987; 316: 661-7.
8. Sturt NJH, Gallagher MC, Bassett P, et al. Evidence
for genetic predisposition to desmoid tumours in
familial adenomatous polyposis independent of the germline
APC mutation. Gut 2004; 53: 1832-6.
9. Clark SK, Neale KF, Landgrebe JC, et al. Desmoids
tumours complicating familial adenomatous polyposis. Br J Surg
1999; 86: 1185-9.
10. Bülow C, Bülow S , Leeds Castle Polyposis Group .
Is screening for thyroid carcinoma indicated in familial
adenomatous polyposis? Int J Colorectal Dis 1997; 12:
240-2.
11. Perrier ND, Van Heerden JA, Goellner JR, et al.
Thyroid cancer in patients with familial adenomatous polyposis.
World J Surg 1998; 22: 738-42.
12. Bilkay U, Erdem O, Ozek C, et al. Benign osteoma
with Gardner syndrome: Review of the literature and report of
a case. J Craniofac Surg 2004; 15: 506-9.
13. Hamilton SR, Liu B, Parsons RE, et al. The molecular
basis of Turcot's Syndrome. N Engl J Med 1995; 332: 839-47.
14. Burt RW, Leppert MF, Slattery ML, et al. Genetic
testing and phenotype in a large kindred with attenuated
familial adenomatous polyposis. Gastroentorology2004; 127:
444-51.
15. Winawer S, Fetcher R, Rex D , et al. (U. S.
Multisociety Task Forece on Colorectal Cancer) Colorectal cancer
screening and surveillance: Clinical guidelines and rationale _
Update based on new evidence. Gastroenterology2003; 124:
544-60.
16. Nugent KP, Phillips RK. Rectal cancer risk in older
patients with familial adenomatous polyposis and an
ileorectal anastomosis: A cause for concern. Br J Surg 1992;1204-6.
17. Ziv Y, Fazio VW , Church JM, Lavery IC, King
TM, Ambrosette p. Stapled ileal pouch anal anastomoses are
safer than handsewn anastomoses in patients with ulcerative colitis.
Am J Surg 1996; 171: 320-3.
18. Rossi BM, Nagakawa, WT, Oliveira FO, Aguiar Jr S, Lopes
ª Câncer de cólon reto e ânus. Tecmed Ed. São Paulo, 2005.
19. Rossi BM, Corvello CM, Anelei A, Epilman C, Paegle
LD, Nagakawa WT, Borghes AY, Amorim C, Duarte APM,
Simpson AJG, Lopes ª Hereditary colorectal tumor: routine case
and multidisciplinary therapeutic approach. South Am J
Câncer 1997; 1: 191-7
Endereço para correspondência:
Prof. Dr. Omar Feres
Departamento de Cirurgia e Anatomia - Disciplina
de Coloproctologia
Hospital das Clínicas da FMRP-USP
email: marlenelucio@yahoo.com.br / feresomar@netsite.com.br
CAMPUS UNIVERSITÁRIO
14.048-900
Recebido em 31/05/2007
Aceito para publicação em 25/06/2007
Trabalho realizado junto à Disciplina de Coloproctologia do Departamento de Cirurgia e Anatomia da Faculdade de Medicina de
Ribeirão Preto-USP (FMRP-USP).